Marjorie Williams
78, Female
Mrs Williams is commenced on IV re-hydration as she has all the signs of dehydration (tachycardia, hypotension, dry mucous membranes). She is given a 250 ml bolus of normal Saline and her blood pressure improves to 110/60. Further IV fluids are prescribed with the aim of making her 2-3 litres positive over the next 24 hours.
- Her Ramipril is stopped.
- She is catheterized to help monitor fluid balance. Her initial urine output is low (10-20 ml/hour)
- She is started on Tazocin (broad spectrum antibiotic) 4.5g every 8 hours
Consider these Questions as to Patient Management in this case
Click on each title below to see the model answer...
Q1. Why does Mrs Williams have a metabolic acidosis?
Model answer:
Metabolic acidosis — The excretion of acid and regeneration of bicarbonate is impaired in the setting of a low glomerular filtration rate (GFR) resulting in metabolic acidosis. Much of the acid that is normally excreted by the kidney is the product of daily metabolism. However, other factors usually contribute to severe acidosis among patients with AKI, who are often critically ill. For example, patients with AKI due to sepsis, trauma, and multi-organ failure often have increased production of lactic acid or ketoacids. Other patients who present with AKI may have metabolic acidosis resulting from loss of bicarbonate or from diarrhea or, less commonly, renal tubular acidosis.
Q2. Why is Mrs Williams’ urine output low when she presents?
Model answer:
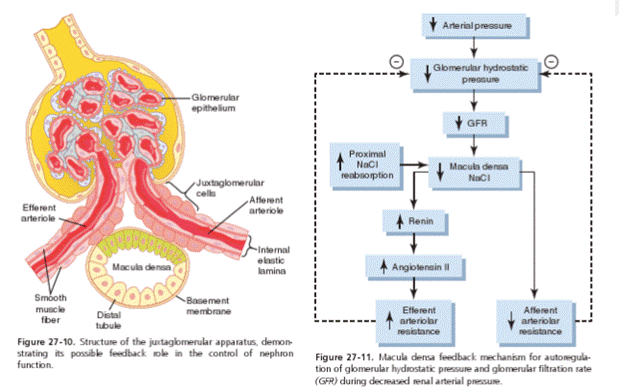
A common causes of prerenal disease is true volume depletion, which includes hypovolemia caused by dehydration, hemorrhage, renal (diuretics) or gastrointestinal (vomiting, diarrhoea) fluid loss leading to decreased renal blood flow. The glomeruli, kidney tubules, and interstitium are intact. Initially the urine volume is typically, but not always, low (oliguria) in prerenal disease due to appropriate increases in both sodium and water reabsorption, which limits further fluid loss. This occurs when cardiac and vascular pressure receptors sense a drop in blood pressure leading to an increase in sympathetic neural tone and the release of both renin (leading to the generation of angiotensin II) and antidiuretic hormone.
With prolonged hypotension and inflammation however, the poorly controlled inflammatory response within the kidney results in widespread renal tubular epithelial dysfunction and a lack of sodium chloride reabsorption in the proximal tubule. This will be detected as an increase in sodium and chloride delivery to the macula densa cells located in the distal tubule. Through tubuloglomerular feedback mechanism this increased tubular NaCl concentration results in widespread vasoconstriction of the afferent arteriole and a subsequent drop in glomerular filtration rate (GFR) that ultimately gives rise to the clinical manifestations of AKI, decreased urine output, and increased serum creatinine which no longer responds to fluid replacement therapy.
Acute tubular necrosis
With prolonged and/or severe ischemia, ATN can occur. This can result in histologic changes, including necrosis, with denuding of the epithelium and occlusion of the tubular lumen by casts and cell debris. This degree of injury can take up to 6 weeks to repair and the patient may need renal replacement therapy to maintain life will kidney repair and regeneration occurs. Recovery of urine output and kidney function will often occur abruptly once tubular repair is complete.
Q3. Why is Mrs Williams hyperkalaemic?
Model answer:
Total body potassium stores are approximately 3000 meq or more (50 to 75 meq/kg body weight) but 98% of this is stored within cells, unlike sodium, which is the major cation in the extracellular fluid and has a much lower concentration in the cells. The intracellular potassium concentration is approximately 140 meq/L compared with 4 to 5 meq/L in the extracellular fluid. The difference in distribution of the two cations is maintained by the Na-K-ATPase pump in the cell membrane, which pumps sodium out of and potassium into the cell in a 3:2 ratio.
The plasma potassium concentration is determined by the relationship between potassium intake, the distribution of potassium between the cells and the extracellular fluid, and urinary potassium excretion. Both cellular uptake and urinary excretion of potassium are impaired in patients with advanced acute or chronic kidney disease.
In normal individuals, dietary potassium is absorbed in the intestines and then largely excreted in the urine, a process that is primarily determined by potassium secretion by the principal cells in the two segments that follow the distal tubule: the connecting segment and cortical collecting tubule (see diagram below). The increase in plasma potassium stimulates the secretion of aldosterone, which directly enhances sodium reabsorption and indirectly enhances potassium secretion.
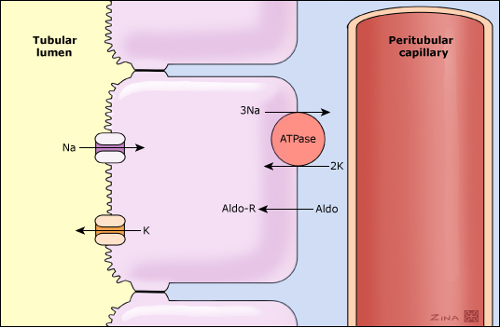
Hyperkalemia is a common complication of acute kidney injury. Even in the presence of normal or increased plasma aldosterone levels, potassium secretion and therefore urinary potassium excretion can be impaired if there is a substantial reduction in sodium and water delivery to the potassium secretory sites in the distal nephron as occurs during decreased tissue perfusion, oliguria and shock. This may be accompanied by other factors such as metabolic acidosis. When acidosis is present buffering of excess hydrogen ions in the cells leads to potassium movement into the extracellular fluid, a transcellular shift that is obligated in part by the need to maintain electroneutrality. The combination of acidosis (causing potassium to leave cells) and reduced potassium excretion (due to reduced sodium delivery to the distal nephron) leads to hyperkalaemia (high serum potassium levels).
Q4. Why was the Ramipril stopped?
Model answer:
Angiotensin converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARBs) reduce systemic blood pressure through inhibiting production of angiotensin II which is a major driver of vasonconstriction. During hypotension this will exacerbate reduction in GFR and renal blood flow. In addition, these drugs specifically inhibit vasoconstriction of the glomerular efferent arteriole thus further reducing glomerular pressure and the glomerular filtration rate. It is therefore recommended that patients on ACEis and ARBs stop taking them when they are acutely unwell or at risk of dehydration.
In addition, ACEi and ARBs inhibit the release of aldosterone thereby reducing sodium reabsorption and preventing potassium excretion in the distal convoluted tubule and collecting ducts.