
Adam Smith
37, Male
You see Adam in clinic with the results of his cardiac MRI scan which does show some scarring in his heart. He is feeling much better on the bisoprolol but is still only on 5mg once a day. He has stopped performing very intense exercise as you advised him to, limiting himself to gentle-to-moderate jogging. However, he still experiences some breathlessness on exertion.
You perform an exercise echocardiogram in clinic which is where echocardiography is performed while Adam is exercising. The aim of this is to look for an inducible outflow tract gradient. As we have discussed above, patients with HCM have left ventricular outflow tract obstruction due to several mechanisms. As with Adam’s case, approximately one-third of patients have a gradient at rest. Another one-third have no gradient at rest. Finally, a further third only develop an inducible gradient, i.e. during exercise, where the heart rate and force of contraction of the heart increase, which can worsen outflow tract obstruction. Therefore sometimes you can see a patient with HCM who is breathless on exertion, but has no outflow tract gradient at rest. In this situation, you have to try and induce and measure an outflow tract gradient during echocardiography by either asking the patient to perform a Valsalva or, even better, performing echocardiography whilst the patient exercises.
Adam’s echocardiogram shows no significant outflow tract gradient at rest (20mmH; we class >30mmHg to be significant and result in symptoms). However, when you re-echo him whilst he is jogging on the treadmill, he develops a gradient of 60mmHg. As a result, you recommend that his bisoprolol dose be increased to 7.5mg once a day and write him a prescription.
Adam’s main concerns are about his risk of SCD and the chances of his children being affected with the condition. These will be addressed in the sections below.
Hypertrophic Cardiomyopathy
Click on the titles of the panels below to open (or close) each section:
Definition
Hypertrophic cardiomyopathy (HCM) is defined as unexplained LV hypertrophy associated with a non-dilated ventricular chamber in the absence of another cardiac or systemic disease that itself would be capable of producing the magnitude of hypertrophy evident in any given patient. The traditional definition requires LVH of ≥15mm to be present for a diagnosis, although theoretically, any magnitude of LVH (≥12mm) may be compatible with the disease. Furthermore, there are a sub-group of patients who carry a genetic mutation for the condition (see below) but have no LVH – the so-called “genotype-positive, phenotype-negative” patients.
Genetics
Hypertrophic cardiomyopathy is the commonest cardiac genetic disorder, with a prevalence of at least 1 in 500 (0.2%) in the general population. It has an autosomal dominant mode of inheritance, meaning that the children of an affected individual each have a 50% chance of inheriting the condition, assuming that the other parent is not affected. Figure 18 below is an example of a pedigree from a family with HCM.
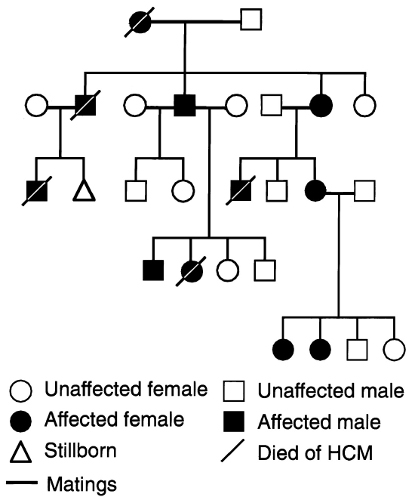
Although most cases of HCM are inherited, a minority may be sporadic, due to a “private” mutation occurring spontaneously (“de novo”) in an individual during development.
The genetic basis of HCM is from mutations in several genes encoding sarcomeric contractile proteins, most commonly beta-myosin heavy chain (gene MYH7) and myosin binding protein C (gene MYBPC). A gene mutation is defined as the permanent alteration of the nucleotide sequence of a gene which is not found in most individuals. Mutations can be disease-causing or innocent (non-pathogenic). The list of known HCM mutations is growing, and we now know of mutations in genes encoding other non-sarcomeric contractile proteins that can cause HCM. A summary of these genes is given in the Figure 19 below. Currently, genetic testing for mutations is routinely performed in 19 genes known to cause HCM or HCM “phenocopies” (conditions that can mimic HCM). Unfortunately, at the moment we have not discovered all the mutations that cause HCM which means that if we gene test a group of people with HCM, at best we will only find a causative gene mutation in 60% of them. Assuming they have good clinical evidence of disease, this does not mean the remaining 40% do not have HCM; it just means they have a mutation we do not yet have knowledge about. With recent advances in molecular genetics, this situation is improving rapidly.
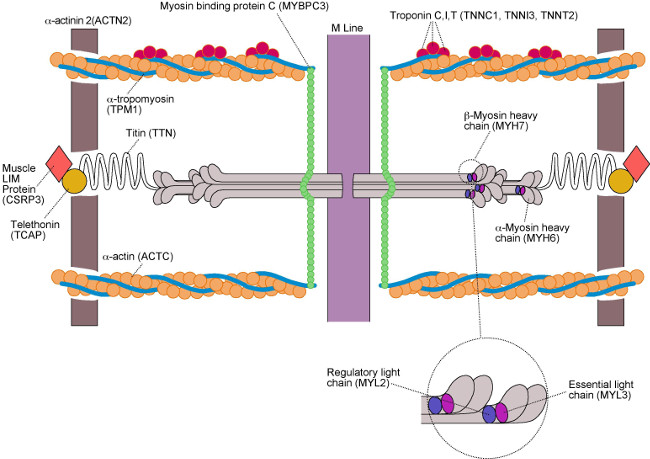
Hypertrophic cardiomyopathy is renowned for its clinical and genetic heterogeneity; this means that within a family, members with the same mutation may have very different clinical disease expression, with some developing severe disease whilst others developing only mild disease or no disease at all (incomplete penetrance). The mechanism behind this is not fully understood, but may be due to “genetic modifiers” which can include anything from other genes to environmental factors, or even other co-existing diseases such as hypertension. Furthermore, HCM also displays variable age-related penetrance, meaning that within the same family, some members may develop disease at an early age, whereas others may not develop disease until later on in life. The clinical implications of this observation are that screening for the condition (see below) in first degree relatives of an affected individual cannot be a “one-off” event; even if no disease is found after screening, this must be repeated routinely throughout a first degree relative’s lifetime. However, genetic testing can help in this situation; if a person with HCM is found to have a positive gene mutation known to cause the disease, predictive genetic testing can be offered to their family members. If the gene test in these family members comes back negative, then further screening will not be required as we know they have not inherited the causative mutation for HCM.
Histopathology
The histological hallmark of HCM is cardiac muscle cell (myocyte) hypertrophy, with the myocytes exhibiting bizarre shapes and being arranged in a chaotic, disorganised pattern which is called “myocyte disarray.” Other features include scar/fibrosis and hypertrophy of the intramural arteriolar cell walls (Figure 20).
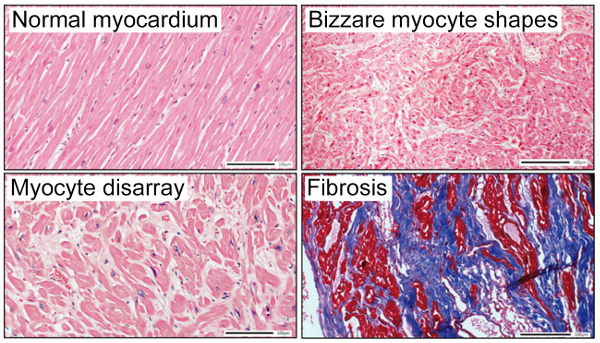
Structural Cardiac Abnormalities
The hallmark of HCM is obviously left ventricular hypertrophy (LVH). This has classically been described as affecting the septum (asymmetric septal hypertrophy, or “ASH”). However, the LVH in HCM can affect any segment of the LV myocardium and occasionally the RV too, and may be present in a variety of different patterns. Indeed, HCM can even produce concentric LVH, which can make it challenging to distinguish HCM from hypertensive heart disease in those patients who have coexisting hypertension. The LVH in HCM usually occurs “at the expense” of the LV cavity (so-called “concentric remodelling”), resulting in some patients with HCM having a small LV chamber size. In addition, the increased stiffness of the heart (caused by LVH, fibrosis and myocyte disarray) results in diastolic dysfunction and increased intra-cardiac pressures. This can result in remodelling of the left atrium and left atrial dilatation.
A discussion and explanation of left ventricular outflow tract obstruction has already been given above in the case scenario. Although SAM of the mitral valve occurs, we now also know that some patients with HCM may exhibit structural abnormalities of the mitral valve and papillary muscles as well, including increases in the size and shape of the mitral valve, elongated mitral valve leaflets and/or redundant tissue, extra chordae tendineae or chordae tendineae with abnormal attachments, and displacement or excessive hypertrophy of the papillary muscles (none of these were seen in Adam). Apart from the mechanisms of SAM described above, these intrinsic mitral valve abnormalities may also contribute to the development of SAM.
Pathophysiology of Symptons
The pathophysiology of outflow tract obstruction, SAM and mitral regurgitation in HCM have already been described and discussed in the case scenario. These may all result in breathlessness, particularly on exertion, and severe outflow tract obstruction may also result in syncope. It is important to remember that not all HCM patients will develop obstruction, as already mentioned above. Diastolic dysfunction, however, contributes to breathlessness and symptoms of heart failure and may be present even in those with non-obstructive disease.
The other mechanism of pre-syncope and syncope in HCM patients are malignant ventricular arrhythmias, namely ventricular tachycardia (VT) and ventricular fibrillation (VF), which arise due to the myocyte disarray and histopathological abnormalities described above. These abnormalities resulting in an unstable myocardial substrate, impairment of electrical impulse transmission, and development of re-entry electrical circuits. Such malignant arrhythmias can cause sudden cardiac death which, for some, may unfortunately be the first manifestation of the disease.
Palpitation in HCM may be caused not only by ventricular arrhythmias (which includes multiple ventricular ectopics in addition to VT and VF), but also supraventricular arrhythmias, particularly atrial fibrillation (AF). Indeed, AF is a cause of much morbidity and mortality in HCM due to stroke, and its presence should result in commencement of anticoagulation with Warfarin regardless of the CHA2DS2-VASc score usually used to risk stratify patients with AF for anticoagulation as this score is not valid in patients HCM. NOACs are not yet licensed in HCM as there in no data on their efficacy in this setting.
Finally, chest pain may occur through a number of mechanisms, but is ultimately due to myocardial ischemia. In the absence of epicardial coronary disease, the mechanisms are thought to be abnormalities of the microvasculature such as arteriolar wall hypertrophy (resulting in microvascular angina) and inadequate capillary density in the face of LVH and a greatly increased LV mass. Severe LV outflow tract obstruction may also contribute to angina in a similar fashion to aortic stenosis.
Clinical Features and Presentation
Many of the clinical features and an example in the way in which patients with HCM can present have already been discussed and described in the case scenario above. Most patients come to medical attention due to symptoms or an incidental finding of a murmur. A further proportion are identified through cascade familial screening of an affected individual with HCM. A smaller proportion are identified fortuitously, for example because of an abnormal ECG performed for another reason e.g. pre-operatively. As mentioned, a small minority present with SCD or aborted (successfully resuscitated) SCD which may be the first (and most feared) presenting symptom.
It should be remembered, of course, that the symptoms and clinical examination findings in HCM will depend both upon severity of disease and, in particular, the presence of left ventricular outflow tract obstruction. Those without outflow tract obstruction may be completely asymptomatic and in these individuals, there may be little to find on examination.
Investigations
The main investigations for patients with HCM have been described above. Patients should generally have a 12-lead ECG, echocardiography, a Holter monitor and exercise test on an annual basis. A cardiac MRI scan should be performed at least once, and may be repeated if disease progression is suspected or in some cases to guide risk stratification as some authorities believe that a high burden of scar or fibrosis is a surrogate risk factor. If an inducible gradient is suspected, then exercise echocardiography is extremely useful, as we have already seen.
As with the general population, patients with HCM are susceptible to coronary artery disease and therefore chest pain of unknown cause should be investigated with a CT coronary angiogram or conventional coronary angiography. In patients with co-existing hypertension in whom LVH is present but who also have clinical features of HCM, then demonstration of LVH despite good blood pressure control on a 24-hour blood pressure monitor can help clarify this diagnostic dilemma. In some patients, a transoesophageal echocardiogram may be required, particularly to evaluate the LV outflow tract and mitral valve architecture in more detail prior to surgical myectomy (see below).
Finally, those patients with large families and young relatives may be offered genetic testing. Although it was previously thought that certain gene mutations produce more severe disease, this theory has now been discounted. Therefore gene testing is mainly useful if a positive result is found, which can then mean other family members can be offered the test to see if they carry the mutation. However, gene testing may also be useful in certain individual cases, where the diagnosis is not clear or in patients who have subtle features of disease and who pose diagnostic dilemmas. Here, a positive gene test can help confirm or clarify the diagnosis. However, because of the ~40% false-negative rate of gene testing in patients with HCM, a negative result in this situation is unfortunately not helpful in ruling out disease.
Management
Management of patients with HCM falls within three categories, as follows.
-
(i) Sympton Control
We have partly discussed symptom control above. The mainstay of treatment for breathlessness due to left ventricular outflow tract obstruction are beta-blockers or verapamil (REMEMBER: patients should only be on one OR the other; combining verapamil and a beta-blocker together is an absolute contra-indication, because the combination can lead to profound heart block and ventricular standstill). Both beta-blockers and verapamil have similar mechanisms of action which have already been described above.
If a patient with outflow tract obstructing is still symptomatic on a top dose of one of these agents, then the Class Ia antiarrhythmic disopyramide can be added in. Disopyramide works predominately through a negative inotropic effect. Finally, in those who remain symptomatic despite full pharmacological therapy, an invasive approach may be required. For those fit for surgery, the gold standard is surgical myectomy where during open heart surgery, the thickened portion of myocardium causing obstruction is cut out (see Figure 21 below). Surgery may also give the opportunity to correct intrinsic mitral valve abnormalities found in HCM patients that can contribute to the development of SAM (see above), such as abnormal chordal attachments. In those not fit for or unwilling to undergo open heart surgery, alcohol septal ablation may be an alternative (see Figure 21 below). During this procedure, alcohol is injected into an artery that supplies the hypertrophied septum, thus causing a localised infarction. With time, the infarcted tissue undergoes necrosis and dies off, causing a reduction in the septal thickness.
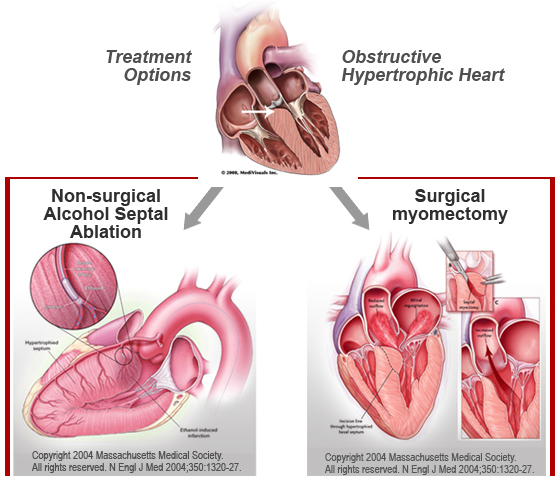
Figure 21: Invasive techniques for management of outflow tract obstruction in HCM Chest pain due to microvascular angina is usually responsive to beta-blockade or verapamil. In those on maximal therapy who still have symptoms, other antianginals such as nitrates may be considered. However, these may not be suitable for those with significant outflow tract gradients as they may exacerbate the gradient due to vasodilatation, so must be used with caution.
Palpitation secondary to arrhythmia is usually readily treatable with beta-blockers or verapamil. As mentioned above, there should be a low threshold to start anticoagulation in patients with evidence of AF, although treatment decisions should be individualised. Patients with bad diastolic dysfunction or outflow tract gradients may tolerate AF extremely poorly, and in these individuals maintaining sinus rhythm with DC cardioversion, amiodarone or an ablation can be a very important aspect of management.
Finally, therapeutic strategies for patients with end-stage heart failure (the “burnt-out” phase of the disease, characterised by wall thinning, chamber dilatation, and reduction in ejection fraction) should with standard heart failure medications including ACE inhibitors or angiotensin receptor blockers, diuretics, and possibly also spironolactone. Severe LV systolic dysfunction is a surrogate risk factor for SCD (see below) and an implantable cardioverter-defibrillator (ICD) may be warranted. In young patients with severe LV dysfunction, cardiac transplantation may be a viable option and they should be sent for assessment to their nearest transplant centre, sooner rather than later.
-
(ii) Risk Stratification and Management of High Risk Patients
Risk stratification is an extremely important aspect of the management of patients with HCM. Traditionally, 5 high-risk features have been described, namely:
- A family history of SCD
- A personal history of unexplained syncope
- Documented non-sustained VT (defined as 3 or more beats at ≥120 beats per minute)
- Severe LVH of≥30mm
- An abnormal blood pressure response to exercise (i.e. failure of systolic blood pressure to increase by ≥25mmHg on exercise and/or a fall in blood pressure on exercise)
In Europe, any 2 of these features would be an indication to implant an ICD for primary prevention. (Of course, if someone presents with aborted SCD or sustained VT, this would automatically be an indication for an ICD regardless of other risk factors).
In recent years, the European Society of Cardiology (ESC) has published an online risk stratification calculator based on a HCM patient’s age, their maximum LV wall thickness on echocardiography, their left atrial size, their maximum left ventricular outflow tract gradient, any family history of SCD, any personal history of non-sustained VT and a personal history of syncope (see “useful links” section below). Some caveats of this calculator are that it is not valid in those ≤16 years of age, in elite or competitive athletes, and in HCM phenocopies.
On the basis of both these methods, Adam qualifies for an ICD: he has 2 traditional risk factors, namely a personal history of syncope and evidence of non-sustained VT on Holter monitoring, and potentially 3 if his family history is taken into account and the deaths attributed to SCD. His risk score using the ESC calculator is very high, with a >14% risk of SCD at 5 years (an ICD is recommended in anyone with a ≥6% risk of SCD at 5 years). Adam is therefore counselled about having an ICD and agrees to be put forward for one.
-
(iii) Family Screening
Finally, all first degree relatives of an individual should undergo screening with history, examination, 12-lead ECG and echocardiography. The timing and frequency of screening depends on the age of the patient. Regular screening is usually commenced in the early teens, around the age of 13, although younger children may also have screening through a paediatrician, especially if they have symptoms. During the teenage years, screening is performed on a yearly basis up till the age of 21, as it is thought that the HCM phenotype is most likely to emerge during the adolescent growth spurt. After the age of 21, screening is recommended on a 3-5 yearly basis, unless symptoms develop in the intervening period, in which case the timing should be brought forward (remember that repeat screening is recommended because of the variable age-related penetrance exhibited by HCM as described in the section on genetics above). As described above, where a positive genetic result has been found in the affected family, predictive genetic testing can be offered to family members to try and negate the need for regular screening if their gene test comes back negative.
Adam is worried about his children so they are referred to a paediatrician for screening and found to have normal ECGs and echocardiograms with no evidence of HCM. Repeat screening is therefore recommended in the early teenage years, unless they develop symptoms before.